During this COVID-19 pandemic the FDA's policy on emergency authorization (EUA) has been a very useful lever in catalysing innovation and investment for accelerating diagnostic tests to the market, but the medical diagnostics industry has now been warned in a recent FDA EUA policy revision that the requirements for the granting of a diagnostic under EUA just became stricter.
Though the policy revision is fairly short at twenty pages for those who want a summary then please consider the following points:
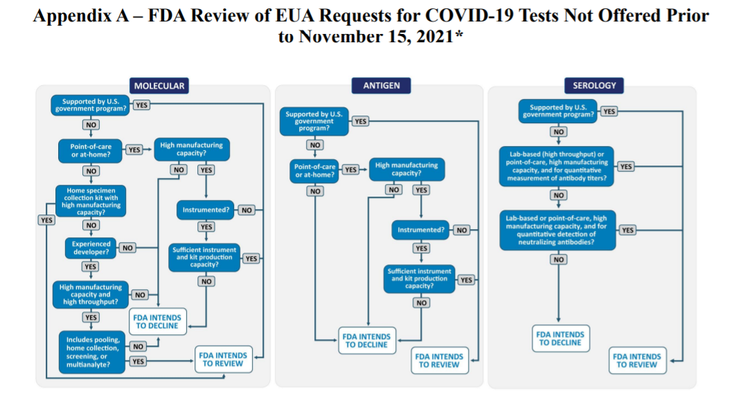
- POINT ONE - If your diagnostic is a point of care device (PoC) and/or can be used at home then the FDA will consider you for EUA, but consider POINT TWO.
- POINT TWO - You must be able to provide convincing evidence that you can manufacture at volumes in excels of 500 k tests per week, within 3-months of the EUA granted.
- POINT THREE -EUA requests for tests which come from or are supported by a US government agency such as Biomedical Advanced Research and Development Authority (BARDA) or the National Institutes of Health’s Rapid Acceleration of Diagnostics (RADx) will be prioritized.
- POINT FOUR - For tests not authorized and not being offered at the time of the publishing of the revised EUA policy, approximately 15 November 2022, then the FDA may only review EUA requests from priority tests, see POINTS ONE, TWO and THREE above.
- POINT FIVE - If the FDA does not receive a confirmation from a test developer that submitted its EUA request prior to February 1 2021 confirming that the developer wants the FDA to continue reviewing its EUA request, then the FDA intends to decline to review, or may decline to further review if the review has already begun.


ABOUT ZP
Zimmer and Peacock is the world's leading ISO13485 contract developer and manufacturer of electrochemical biosensors and IVDs. Our services include CE marking and FDA submissions for the devices we develop and manufacture with our partners.